
DNA double-strand break repair
Simultaneous breaks in both strands of the DNA duplex are potentially lethal events that can lead to permanent loss of genetic information as well as many different types of misrepair products. These types of events can act as initiating lesions in cancer and can also generate recombination products that determine the outcome of cancer progression. We are interested in the mechanisms underlying the repair of DNA double-strand breaks, particularly the initial events that occur at a break site within a few seconds to a few minutes of break formation. One of the key regulators that binds to double-strand breaks and coordinates its repair is the Mre11/Rad50/Nbs1 (MRN) complex, an ancient molecular machine that processes DNA ends, pairs the broken ends together, and participates in signaling the presence of breaks within cells (1–6). We study the activities of recombinant MRN complexes in eukaryotic cells and also in vitro to characterize its actions on different types of DNA substrates and recombination intermediates.
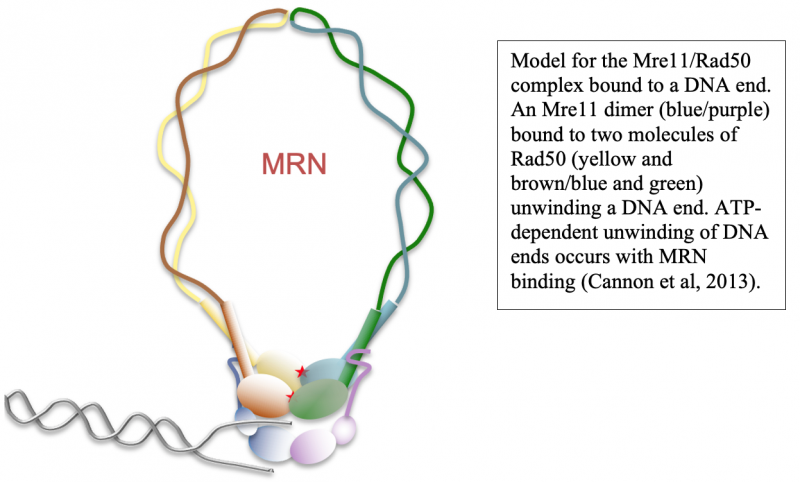
DNA end resection, the removal of hundreds of nucleotides from the 5' strand at double-strand breaks, is an essential part of repair that promotes homologous recombination. Through our work over the past several years we have shown that the MRN complex plays a number of important roles in this process, including the initial short-range resection of the 5ʹ strand by Mre11, recruitment of long-range 5ʹ to 3ʹ nucleases, and displacement/removal of the Ku heterodimer that also binds rapidly to DNA ends and promotes non-homologous end joining (4, 7–9).
Although homologous recombination and non-homologous end joining have long been thought to be competing pathways of DNA repair, our recent results show that the MRN complex works cooperatively with Ku and its binding partner DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to recognize and process ends. This sequential model of DNA end processing is consistent with our results and that of many groups in the field and suggests that non-homologous end joining is attempted first, followed by end processing for homologous recombination (10). Recent work from the lab has also established a novel chromatin immunoprecipitation method to isolate the DNA fragments generated by MRN at DNA-PK-bound sites in mammalian cells, which we have used to validate and extend our in vitro findings (9). Current studies are investigating the patterns, regulation, and cell cycle specificity of these processing events and how MRN-dependent DNA processing occurs in human cells during physiologically relevant DNA damage exposure.
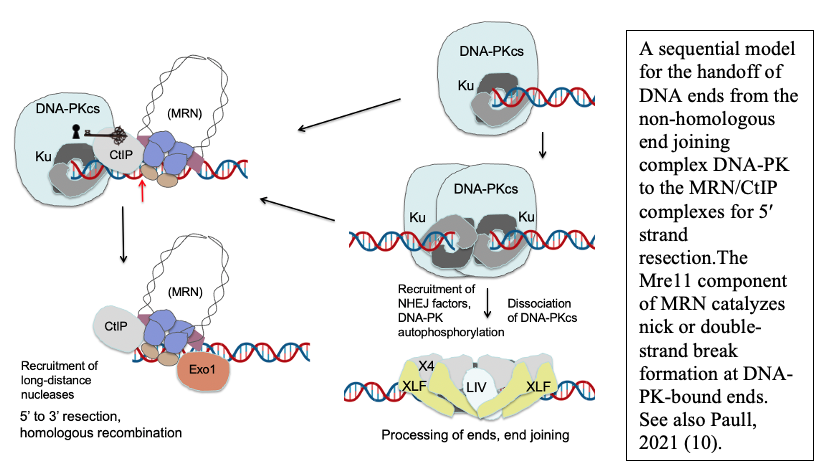
DNA end processing by the MRN complex also requires stimulation by the CtIP protein, a factor specific to eukaryotic cells that is phosphorylated in a cell cycle-dependent manner and is critical for the activation of MRN processing. We have shown that the CtIP protein and its phosphorylation are essential for the activity of Mre11 at DNA-PK-bound sites, both in a reconstituted purified system and in human cells (9).
Transcription-associated DNA lesions
CtIP is a multi-functional protein that also plays important roles at sites of DNA damage that overlap with actively transcribed genes. We have found that loss of CtIP or of Sae2, the ortholog of CtIP in budding yeast, generates R-loops—3-stranded structures containing RNA-DNA hybrids and a displaced DNA strand (11). How CtIP prevents transcription-associated DNA damage is not yet understood, but this is an area of active research in our group. Current studies are investigating how the nuclease activity of CtIP affects the processing of R-loop structures in human cells and what other factors are at R-loops that promote their recognition and removal.
DNA Damage Signaling
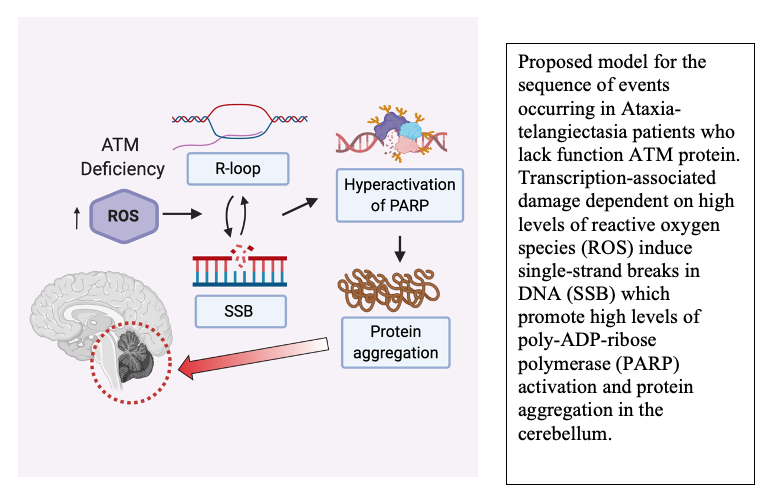
The MRN complex works in concert with the Ataxia-Telangiectasia-Mutated (ATM) protein kinase that phosphorylates many downstream targets responsible for checkpoint activation and DNA damage signaling in eukaryotes. We have previously shown that MRN recruits ATM to broken DNA ends and activates its kinase activity at these sites (12, 13), initiating a cascade of phosphorylation that ultimately affects hundreds of targets in the nucleus. Loss of ATM function in humans is the cause of the genetic disorder Ataxia-Telangiectasia (A-T), which results in a diverse set of clinical symptoms that include childhood-onset cerebellar ataxia, cancer predisposition, and immunodeficiency. Despite many years of study, the molecular basis of neurodegeneration in A-T is not understood. Our recent results examining cerebellum tissue from A-T patients and controls showed that one of the common features of patient brain tissue is protein aggregates—irreversible, detergent-resistant protein assemblies that are found in many common neurodegenerative diseases (14). From experiments in human cells in culture, we know that these aggregates are dependent on the accumulation of transcription-dependent single-strand breaks and that high levels of reactive oxygen species in ATM-deficient cells contributes to these events (15).
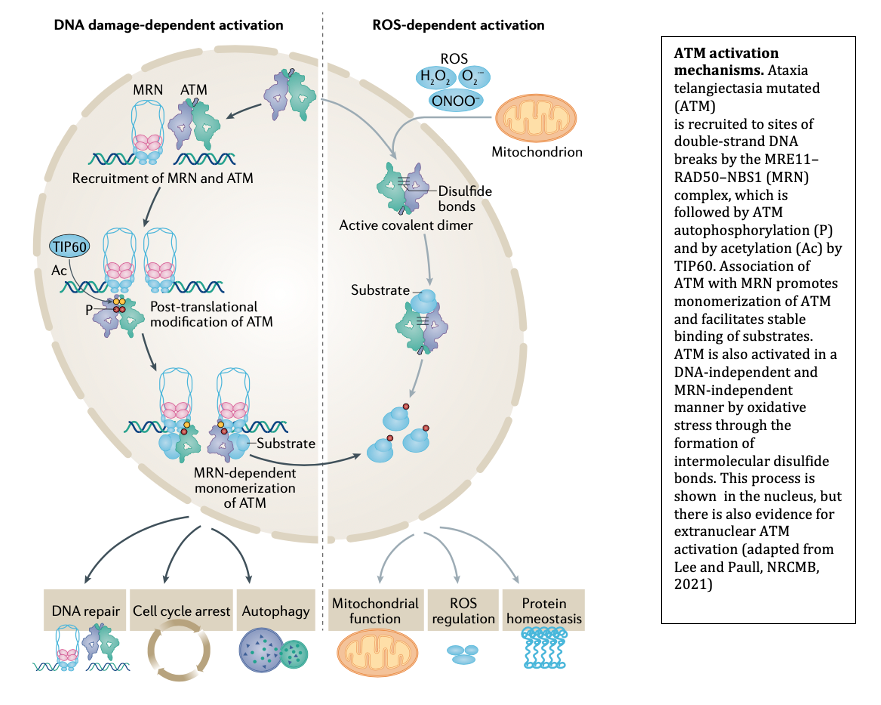
Oxidative Stress Responses
We previously discovered that ATM can be activated in an MRN-independent manner through direct oxidation (16). This pathway is important for cellular control of antioxidant functions and for global responses of human cells to reactive oxygen species. Consistent with this role, cells from A-T patients exhibit signs of chronic oxidative stress and some of the tissues that are most affected by ATM loss in mouse models of A-T can be protected from damage through exposure to antioxidants. Importantly, one of the mutations in the ATM gene that causes early-onset ataxia specifically eliminates the ROS-dependent ATM signaling pathway (R3047X), suggesting that loss of this pathway is sufficient to generate the neurodegeneration that occurs in this disease. Current studies are aimed at understanding the molecular basis of the DNA damage that occurs in ATM-deficient cells and how this generates protein aggregates that are associated with and may play a causal role in the neurodegeneration process.
Poly-ADP-ribosylation and DNA damage-induced loss of protein homeostasis
Another very rapid response to DNA damage is the binding and activation of poly-ADP-ribose (PAR) polymerases that catalyze formation of PAR chains on proteins at damage sites. We know from analysis of A-T patient tissues that PAR levels are high in A-T cerebellum tissue, and from experiments in cells in culture we know that polymerization of PAR promotes the protein aggregation events that occur in the absence of ATM function (14). PAR polymers are similar to nucleic acids in charge but can be made and degraded dynamically by PARP enzymes and PAR glycohydrolases. Based on our recent results in patient tissue and human neurons differentiated in culture, we postulate that DNA damage occurring due to high levels of reactive oxygen species in ATM-deficient neurons promotes chronic hyperactivation of PARP enzymes and ultimately results in the irreversible loss of protein function that manifests as protein aggregation (17). Current studies in the lab seek to understand the molecular mechanisms underlying these events and to further understand the changes occurring in human A-T patients that are responsible for the childhood-onset neurodegeneration in this disease.
Measurements of chaperone clients in human cells
There are significant effects of DNA damage on protein homeostasis but we do not understand these at a molecular level. One way to interrogate misfolded proteins and protein aggregates in cells is by quantifying proteins bound to the ubiquitous protein chaperone HSP70. We have generated a ubiquitination-based system for identifying HSP70 chaperone clients in human cells and have shown that it can be a powerful readout of changes in components in multi-subunit complexes as well as the effects of unstable protein expression (18).
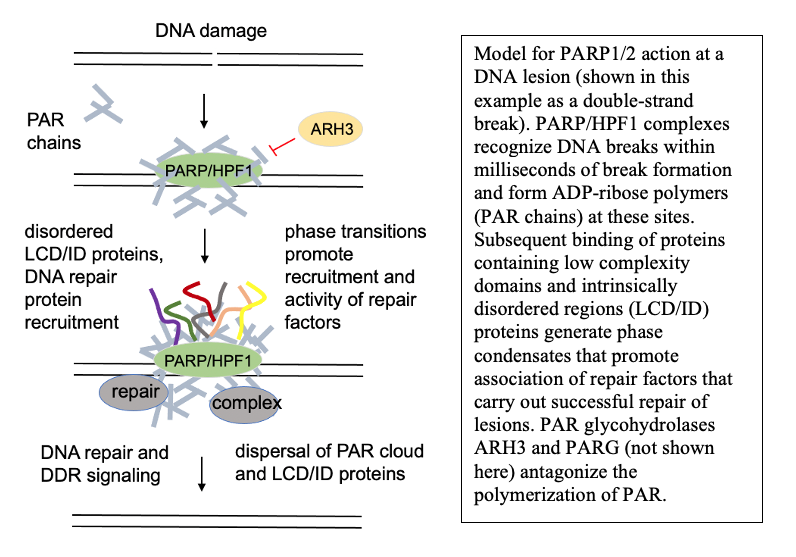
Poly-ADP-ribosylation and DNA repair outcomes
Under normal conditions, PARP activation at sites of DNA lesions promotes successful DNA repair. One of the consequences of this activation is the formation of PAR-dependent phase condensates—protein assemblies seeded by PAR that are dependent on nucleic acid binding domains. Currently we know relatively little about the function of DNA damage-dependent phase condensates and how they function in DNA repair outcomes, but we hypothesize that the condensates are an essential component of DNA repair in living cells. Our studies are focusing on this question, using both in vitro reconstituted systems and human cells to address the molecular basis of these relationships.
1. T. T. Paull, 20 Years of Mre11 Biology: No End in Sight. Mol. Cell. 71, 419–427 (2018).
2. B. Cannon, J. Kuhnlein, S. H. Yang, A. Cheng, D. Schindler, J. M. Stark, R. Russell, T. T. Paull, Visualization of local DNA unwinding by Mre11/Rad50/Nbs1 using single-molecule FRET. Proceedings of the National Academy of Sciences of the United States of America. 110, 18868–73 (2013).
3. R. A. Deshpande, G. J. Williams, O. Limbo, R. S. Williams, J. Kuhnlein, J.-H. Lee, S. Classen, G. Guenther, P. Russell, J. A. Tainer, T. T. Paull, ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 33, 482–500 (2014).
4. R. A. Deshpande, J.-H. Lee, S. Arora, T. T. Paull, Nbs1 Converts the Human Mre11/Rad50 Nuclease Complex into an Endo/Exonuclease Machine Specific for Protein-DNA Adducts. Mol. Cell. 64, 593–606 (2016).
5. R. A. Deshpande, J. H. Lee, T. T. Paull, Rad50 ATPase activity is regulated by DNA ends and requires coordination of both active sites. Nucleic acids research (2017), doi:10.1093/nar/gkx173.
6. J. H. Lee, M. R. Mand, R. A. Deshpande, E. Kinoshita, S. H. Yang, C. Wyman, T. T. Paull, Ataxia Telangiectasia-Mutated (ATM) Kinase Activity Is Regulated by ATP-driven Conformational Changes in the Mre11/Rad50/Nbs1 (MRN) Complex. J Biol Chem. 288, 12840–51 (2013).
7. M. L. Nicolette, K. Lee, Z. Guo, M. Rani, J. M. Chow, S. E. Lee, T. T. Paull, Mre11-Rad50-Xrs2 and Sae2 promote 5’ strand resection of DNA double-strand breaks. Nat Struct Mol Biol. 17, 1478–85 (2010).
8. L. R. Myler, I. F. Gallardo, M. M. Soniat, R. A. Deshpande, X. B. Gonzalez, Y. Kim, T. T. Paull, I. J. Finkelstein, Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol Cell. 67, 891-898 e4 (2017).
9. R. A. Deshpande, L. R. Myler, M. M. Soniat, N. Makharashvili, L. Lee, S. P. Lees-Miller, I. J. Finkelstein, T. T. Paull, DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci Adv. 6, eaay0922 (2020).
10. T. T. Paull, Reconsidering pathway choice: a sequential model of mammalian DNA double-strand break pathway decisions. Curr Opin Genet Dev. 71, 55–62 (2021).
11. N. Makharashvili, S. Arora, Y. Yin, Q. Fu, X. Wen, J.-H. Lee, C.-H. Kao, J. W. Leung, K. M. Miller, T. T. Paull, Sae2/CtIP prevents R-loop accumulation in eukaryotic cells. Elife. 7 (2018), doi:10.7554/eLife.42733.
12. J. H. Lee, T. T. Paull, Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 304, 93–6 (2004).
13. J.-H. Lee, T. T. Paull, ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 308, 551–554 (2005).
14. J.-H. Lee, S. W. Ryu, N. A. Ender, T. T. Paull, Poly-ADP-ribosylation drives loss of protein homeostasis in ATM and Mre11 deficiency. Mol Cell. 81, 1515-1533.e5 (2021).
15. J.-H. Lee, M. R. Mand, C.-H. Kao, Y. Zhou, S. W. Ryu, A. L. Richards, J. J. Coon, T. T. Paull, ATM directs DNA damage responses and proteostasis via genetically separable pathways. Sci Signal. 11 (2018), doi:10.1126/scisignal.aan5598.
16. Z. Guo, S. Kozlov, M. F. Lavin, M. D. Person, T. T. Paull, ATM activation by oxidative stress. Science. 330, 517–521 (2010).
17. J.-H. Lee, T. T. Paull, Cellular functions of the protein kinase ATM and their relevance to human disease. Nat Rev Mol Cell Biol (2021), doi:10.1038/s41580-021-00394-2.
18. S. W. Ryu, R. Stewart, D. C. Pectol, N. A. Ender, O. Wimalarathne, J.-H. Lee, C. P. Zanini, A. Harvey, J. M. Huibregtse, P. Mueller, T. T. Paull, Proteome-wide identification of HSP70/HSC70 chaperone clients in human cells. PLoS Biol. 18, e3000606 (2020).