
Molecular Dynamics Simulations
Protein Electrostatics
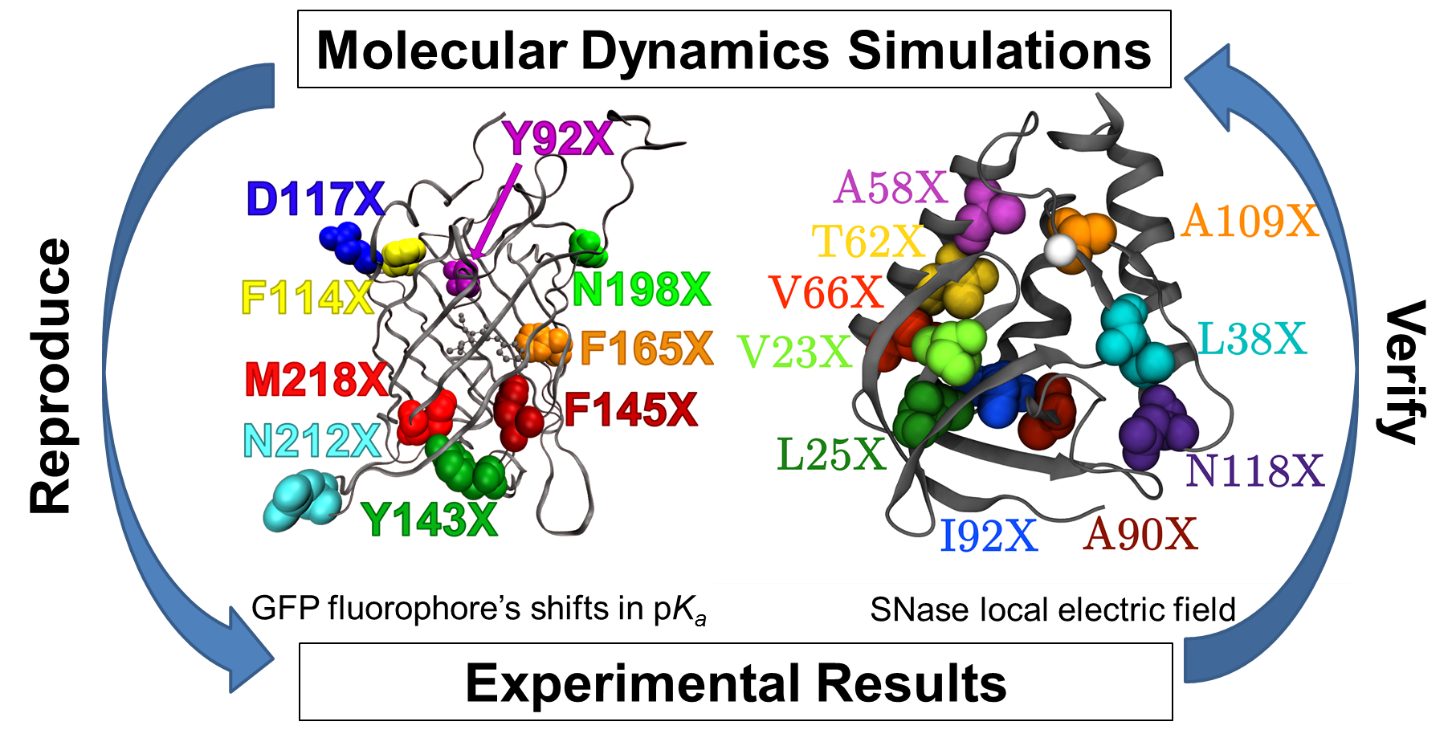
We use molecular dynamics (MD) simulations, coupled with electrostatic calculations, to interpret our experimental results. This allows us to test and refine computational methodologies directly against our experimental data. Once we gain confidence in a simulation strategy, this allows us to design new experiments that test specific hypotheses. In this way, both experimental understanding and modeling ability grow collaboratively to enhance our molecular-level understanding of difficult problems in biolmolecular structure, dynamics, and function. This strategy is central to our investigations of every protein and peptide complex of interest to our laboratory, including Ras and other members of the GTPase superfamily, GFP, and SNase.
Protein-surface interactions
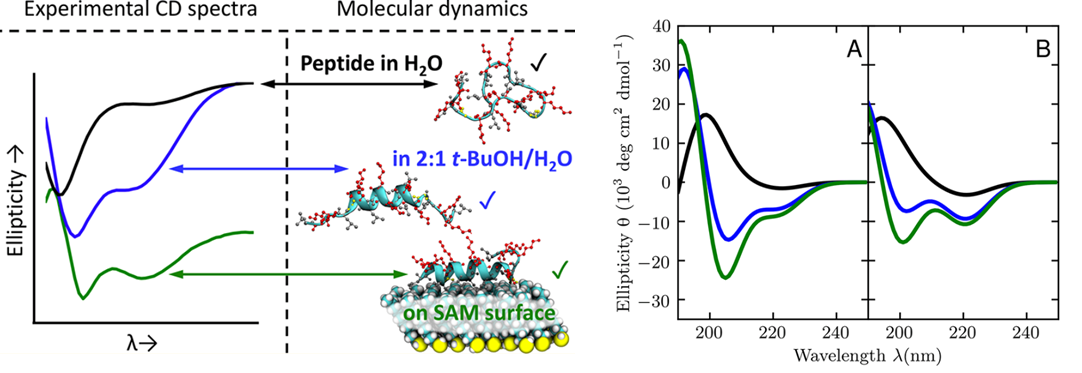
One reason that successfully immobilizing functional proteins on inorganic surfaces is so challenging is because of the lack of understanding of the effect of nonaqueous environments on protein structure from both experimental and computational perspectives. Because most experimental information about protein structure comes from the Protein Data Bank and is collected from an aqueous solvent environment, modern force fields for molecular dynamics (MD) simulations are parameterized against these data. The applicability of such force fields to biomolecules in different environments, including when in contact with surfaces and substrates, must be validated. We use our experimental results to test quantitative agreement between experiments and simulations of solvent- and surface-induced conformational changes of a positively charged peptide in these three environments. For example, we have shown that a surface-bound peptide must fold before chemically reacting with the surface. These advances are guiding further simulations of peptides and proteins in diverse and complex environments.